AlphaFold – AI Meets Proteins
#HumanDataDeep Mind solves a 50 years old biology problem.
Biology is a vast and complex field in which several problems has not been resolved at this day. However, a great scientific discovery has just been carried out thanks to the DeepMind company specializing in artificial intelligence.
Introduction
This year may have been one of the worst in human history, but it is coming to an end on a positive note for life sciences. Recently, Google’s DeepMind announced that they have developed an AI algorithm that officially cracked a long standing problem in biology, in what could be called a major breakthrough in scientific research.
The problem in question, which is referred to as the Protein Folding Problem, is about the folding process that proteins go through once synthesized and the final 3D shapes they form in the space. Solving this challenge is of great importance since a better understanding of proteins and their structures would have major implications in many scientific disciplines such as bioinformatics, biochemistry, genetic engineering, molecular biology and medicine, to cite a few.
“These algorithms are now becoming mature enough and powerful enough to be applicable to really challenging problems”, said DeepMind’s founder and CEO. This marks an important milestone in computational biology and should accelerate research and scientific discovery in all life sciences. So what is the protein folding problem ? How did DeepMind solve it ? And what are the implications of this discovery ?
Protein Folding Problem
Proteins are one of the major classes of biological macromolecules and are essentially made of a sequence of chemicals known as amino-acids. These sequences are assembled according to the genetic instructions of the DNA. Proteins are present in all living creatures where they play a key role in all tasks and chemical processes essential to sustaining life and vital for bodily functions. Examples include regulating pH levels, defending the body against foreign invaders (Antibodies), acting as a messenger between cells (Hormonal Proteins), enabling muscle contraction and movement (Contractile Proteins), converting food into energy (Enzymes) etc. Given their important role, they are regarded as the basic unit of life, and developing a deeper knowledge of their structure and functions can help researchers gain a better understanding of living organisms and decode the machinery of life.
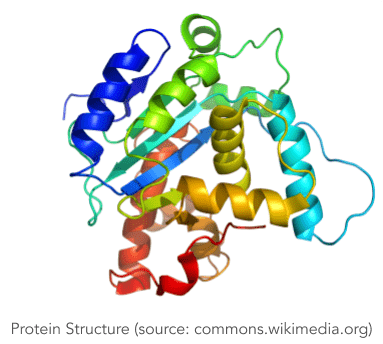
As soon as they are created, proteins fold up rapidly in an infinite number of ways until they reach a stable three dimensional structure referred to as the Native State, which holds the key to the functions that the protein will carry out afterwards. The number of possible shapes is essentially endless, but to make matters easier, it is often assumed that the folding process obeys the Thermodynamic Principe, which means that the Native State is characterized by a minimum level of Gibbs Free Energy.
Knowledge of the Native State is very important because diseases are linked to the roles of proteins in catalyzing chemical reactions (Enzymes), fighting disease (Antibodies) etc. “Even tiny rearrangements of these vital molecules can have catastrophic effects on our health, so one of the most efficient ways to understand disease and find new treatments is to study the proteins involved,” said Dr John Moult of the University of Maryland, US.
The Protein Folding Problem, also known as the Protein Structure Prediction, aims to predict the Native State of a protein (its final 3D shape) based on the chain of amino-acids that make up said protein. The major underlying assumption here is that the type and order of amino-acids in the string, contains all the information necessary to predict the 3D shape. This results from the observation that interactions between the 20 different types of amino-acids are responsible for steering the folding process, thereby forming the different low-level patterns (curls, loops, pleats …) that make up the protein’s final structure. As an example, older studies on the protein folding problem classified amino-acids solely based on their hydrophobicity after finding that attraction to water constitutes one of the major forces of interaction that influences close to 70% of the folding behavior.
Conceptually, we can assume the existence of a mapping between the input space of all proteins, or equivalently, the space of all possible sequences of amino-acids, and the output space of all possible 3D shapes. The goal of the Protein Folding Problem is to approximate this unknown function. Given the massive size of the search space, this is a difficult optimization problem that was proved to be NP-complete. Many promising techniques were proposed in the scientific literatures and most of them are either based on genetic algorithms or reinforcement learning.
Today, there are more than 200 millions known proteins in humans but also in other species such as bacteria and viruses, but scientists have only managed to uncover the structure of a small fraction of them. Traditionally, these 3D shapes are determined through a long manual process that requires an extensive amount of time, expensive lab equipments and scarce human expertise, making the solution simply not scalable. This is the main reason the research community turned to computational methods for a chance at solving this long standing problem that puzzled researchers for half a century.
AlphaFold
DeepMind is a London based company and interdisciplinary research laboratory specialized in artificial intelligence. It rose to fame after developing a neural network that learned how to play Atari video games to a level that rivaled human players. Later in 2016, the company made headlines after their novel AI program, AlphaGo, won against the world champion in the popular strategy board game called Go.
As it turns out, human level gameplay was never intended to be the main goal of DeepMind. From the company’s standpoint, games just provided an ideal training ground for AI technology to reach a certain level of maturity before it can be effectively used on real-world problems.
In 2018, the company proposed AlphaFold, its first attempt at solving the Protein Folding Problem which ended up placing first in the CASP13 international competition. CASP (Critical Assessment of Techniques for Protein Structure Prediction) is a community forum that was created in 1994 by scientists interested in the protein folding problem. The goal is to allow researchers to share their ideas and advances on the topic. To that end, the community holds a competition every two years where scientists and research groups can participate in order to test the performance of their methods in solving the problem against real experimental data. Specifically, entrants are given the sequences of 100 proteins for which the structures have been previously determined based on lab work, but not announced publicly. Participants use their computational methods to predict the shapes of these proteins, which are subsequently compared to their “true” structures in order to compute a performance score.
This year, DeepMind proposed its second version of AlphaFold, which is significantly different from the first iteration in terms of architecture. This time around, the researchers leveraged attention and transformer mechanisms combined with working ideas from the first version. The algorithm was trained on a public dataset, the Protein Data bank, containing roughly 170,000 protein sequences and their corresponding structures (i.e. labels). The training was carried out on hardware comprising 122 3rd generation TPUs (special chips tailor made to accelerate deep learning training), which is equivalent to 100-200 modern GPUs and lasted for a few weeks. The company declined to reveal the cost of training, but for the sake of reference, Google charges $32/hour for a single 3rd generation TPU which results in about $690,000 for a single week. AlphaFold v2 participated in CASP14 (this year’s competition), and won 1st place for the second time in a row. The second version of the AI program achieved a median score of 92.5/100, with 90 being the equivalent to traditional experimental methods. As a result, CASP organizers and DeepMind researchers declared the protein folding problem to be solved.
Implications
The ability to investigate the shape of proteins quickly and accurately has the potential to revolutionize life sciences. Since the 3D structure of a protein determines its role and functions, then predicting the outcome of the folding process using a computer means that researchers can uncover what each protein does. This can typically allow them to answer questions such as how antibodies fight viruses and how insulin regulates sugar levels.
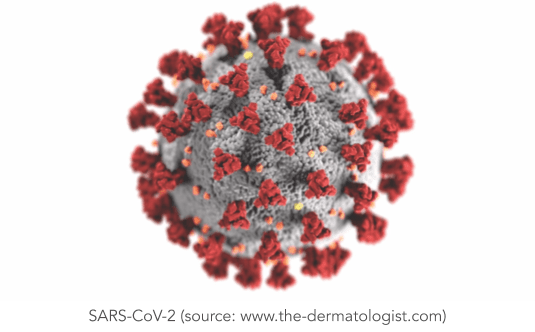
Protein structures also play a pivotal role in drug design and discovery and in understanding health and the mechanisms that drive some diseases, such as cancer. It can take years of research and billions of dollars in investments to create a new drug, which inevitably holds back fundamental research efforts. Predicting the structures of proteins related to a disease has the potential to accelerate research and immensely reduce the associated cost. An example that perhaps everyone can relate to is the Corona virus. When Covid-19 was brought to light, we knew very little about it. SARS-CoV-2, the virus responsible for Covid-19, is made of 30 different types of proteins, third of which are poorly understood. In particular, scientists have been studying the interaction between the spike protein found on the surface of the SARS-CoV-2 virus and receptors in human cells. To help advance our understanding of the virus, the research team behind AlphaFold used their AI program to predict the structures of a few understudied proteins associated with the Corona virus.
Furthermore, limited knowledge of protein structures and lack of funding has significantly hindered progress in understanding some tropical diseases that impact the lives of millions and result in many deaths every year. DeepMind said that it started working with a few research groups to focus their efforts on some of these diseases.
Solving the protein folding problem should also pave the way for new opportunities to emerge such as finding “green enzymes” that break down plastic waste thereby reducing pollution, developing more nutritious crop to improve human health, or efficiently capturing carbon from the atmosphere.
What’s Next?
AlphaFold v2 is definitely a promising news for the scientific community. However, as with any other research effort, improvements can be made. The research team behind the algorithm believes that it can be further enhanced to be even more accurate. Also, the only benchmark we have so far is the score on the test sample provided by the CASP community. As much as we want to believe in this achievement, further testing remains necessary to better understand the limitations of this predictive model, and accurately assess its generalization capabilities.
Now that the problem is arguably solved for single proteins, the next logical step is developing new techniques to predict the shape of protein complexes, which are structures composed of two or more proteins and form the cornerstone of many biological processes. Another knowledge gap is figuring out how proteins interact with other macromolecules such as lipids, carbohydrates or DNA.