


AlphaFold : L’IA au service de la biologie
#HumanDataDeepMind résout un problème de biologie de 50 ans.
La biologie est un domaine vaste et complexe dans lequel plusieurs problèmes sont encore non résolus à ce jour. Une formidable découverte scientifique vient toutefois d’être effectuée grâce à l’entreprise DeepMind spécialisée dans l’intelligence artificielle.
Introduction
Cette année est peut-être l’une des pires de l’histoire de l’humanité, mais elle s’achève sur une note positive pour les sciences de la vie. Récemment, la filiale DeepMind de Google a annoncé la mise au point d’un algorithme d’IA qui a officiellement résolu un problème de longue date en biologie, marquant ainsi une percée majeure dans la recherche scientifique.
Le problème en question, appelé le “Repliement des Protéines”, concerne le processus physique de repliement suivi par les protéines une fois synthétisées, avant d’atteindre leurs structures tridimensionnelles finales dans lesquelles elles sont fonctionnelles. La résolution de ce problème est d’une grande importance car une meilleure compréhension des protéines et de leurs structures aurait des implications majeures dans de nombreuses disciplines scientifiques telles que la bioinformatique, la biochimie, le génie génétique, la biologie moléculaire et la médecine, pour n’en citer que quelques-unes.
« Ces algorithmes deviennent maintenant suffisamment matures et puissants pour être applicables à des problèmes vraiment difficiles », a déclaré le fondateur et le PDG de DeepMind. Cela marque une étape importante dans le domaine de la biologie computationnelle et devrait accélérer la recherche et les découvertes scientifiques dans toutes les sciences de la vie. Quel est donc le problème du repliement des protéines ? Comment DeepMind l’a-t-il résolu ? Et quelles sont les implications de cette découverte ?
Le problème du repliement des protéines
Les protéines sont considérées comme l’une des principales classes de macromolécules biologiques et sont essentiellement constituées d’une séquence de substances chimiques connues sous le nom d’acides aminés. Ces séquences sont assemblées selon les instructions génétiques de l’ADN. Les protéines sont présentes dans tous les êtres vivants où elles jouent un rôle clé dans les tâches et les processus chimiques essentiels au maintien de la vie et aux fonctions corporelles. Elles régulent par exemple le niveau du pH, défendent le corps contre les envahisseurs étrangers (anticorps), agissent comme messagers entre les cellules (protéines hormonales), permettent la contraction et le mouvement des muscles (protéines contractiles), transforment les aliments en énergie (enzymes) etc. Compte tenu de leur rôle important, elles sont considérées comme l’unité de base de la vie, et le développement d’une connaissance plus approfondie de leurs structures et de leurs fonctions peut aider les chercheurs à mieux comprendre les organismes vivants et à décoder les mécanismes de la vie.
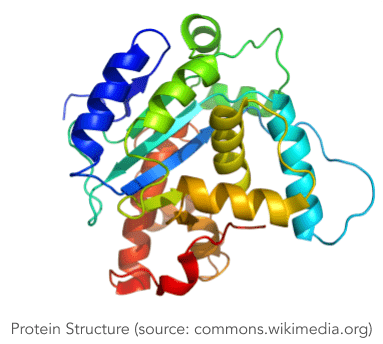
Dès qu’elles sont créées, les protéines se replient rapidement et de différentes manières jusqu’à ce qu’elles atteignent une structure tridimensionnelle stable appelée « État Natif », qui détient la clé des fonctions que la protéine va ensuite remplir. Le nombre de formes possibles est essentiellement infini, mais pour faciliter les choses, on suppose souvent que le processus de repliement obéit au principe thermodynamique, ce qui signifie que l’état natif est caractérisé par un niveau minimum d’énergie libre de Gibbs.
La connaissance de l’état natif est très importante car les maladies sont liées au rôle des protéines dans la catalyse desréactions chimiques (enzymes), la lutte contre les maladies (anticorps), etc.
« Même de minuscules réarrangements de ces molécules vitales peuvent avoir des effets catastrophiques sur notre santé, c’est pourquoi l’un des moyens les plus efficaces de comprendre les maladies et de trouver de nouveaux traitements est d’étudier les protéines impliquées », a déclaré le Dr John Moult de l’université du Maryland, aux États Unis.
Le Protein Folding Problem, également connu sous le nom de Protein Structure Prediction, vise à prédire l’état natif d’une protéine (sa structure 3D finale) en se basant sur la chaîne d’acides aminés qui composent ladite protéine. La principale hypothèse sous-jacente ici est que le type et l’ordre des acides aminés dans la chaîne contiennent toutes les informations nécessaires pour prédire la forme 3D. Ceci résulte de l’observation que les interactions entre les 20 différents types d’acides aminés sont responsables du pilotage du processus de repliement, formant ainsi les différents motifs de bas niveau (boucles, plis…) qui constituent la structure finale de la protéine. Par exemple, des études plus anciennes sur le problème du repliement des protéines classaient les acides aminés uniquement en fonction de leur hydrophobicité après avoir constaté que l’attraction de l’eau constitue l’une des principales forces d’interaction qui influence près de 70% du comportement de repliement.
Conceptuellement, nous pouvons supposer qu’il existe une fonction de correspondance entre l’espace de toutes les protéines, ou de manière équivalente, l’espace de toutes les séquences possibles d’acides aminés, et l’espace de toutes les structures 3D possibles. L’objectif du problème du repliement des protéines est de trouver une approximation de cette fonction inconnue. Étant donné la taille massive de l’espace de recherche, il s’agit d’un problème d’optimisation difficile qui s’avère être NP-complet. De nombreuses techniques prometteuses ont été proposées dans la littérature scientifique et la plupart d’entre elles sont soit basées sur des algorithmes génétiques, soit sur l’apprentissage par renforcement.
Aujourd’hui, il existe plus de 200 millions de protéines connues chez l’homme mais aussi chez d’autres espèces comme les bactéries et les virus, mais les scientifiques n’ont réussi à découvrir la structure que d’une petite fraction d’entre elles. Traditionnellement, ces formes 3D sont déterminées à travers un long processus manuel qui nécessite beaucoup de temps, des équipements de laboratoire coûteux et une expertise humaine rare, ce qui rend la solution tout simplement impossible à appliquer à l’échelle. C’est la raison principale pour laquelle la communauté des chercheurs s’est tournée vers les méthodes informatiques pour avoir une chance de résoudre ce problème de longue date qui a intrigué les chercheurs pendant un demi-siècle.
AlphaFold
DeepMind est une entreprise et un laboratoire de recherche interdisciplinaire basé à Londres et spécialisé dans l’intelligence artificielle. Elle a atteint la célébrité après avoir développé un réseau de neurones qui a appris à jouer aux jeux vidéo Atari à un niveau rivalisant avec celui des humains. Plus tard, en 2016, la société a fait la une des journaux après que son nouveau programme d’IA, AlphaGo, ait remporté une victoire contre le champion du monde dans le jeu de stratégie populaire appelé Go.
Atteindre le niveau human dans des jeux vidéo n’a jamais été l’objectif principal de DeepMind. Du point de vue de la société, les jeux vidéo constituent simplement un terrain d’entraînement idéal pour que les technologies de l’IA atteignent un certain
niveau de maturité avant de pouvoir être utilisée efficacement pour résoudre des problèmes du monde réel.
En 2018, la société a proposé AlphaFold, sa première tentative pour résoudre le problème du repliement des protéines, qui a fini par se classer en première place dans la compétition internationale CASP13. CASP (Critical Assessment of Techniques for Protein Structure Prediction) est un forum communautaire qui a été créé en 1994 par des scientifiques intéressés par le problème du repliement des protéines. L’objectif est de
permettre aux chercheurs de partager leurs idées et leurs avancées sur le sujet. Dans ce cadre, la communauté organise tous les deux ans un concours auquel les scientifiques et les groupes de recherche peuvent participer afin de tester la performance de leurs méthodes dans la résolution du problème par rapport à des données expérimentales réelles. Plus précisément, les participants reçoivent les séquences d’acides aminés de 100 protéines dont les structures ont été préalablement déterminées sur la base de travaux de laboratoire, mais qui n’ont pas été annoncées publiquement. Les participants utilisent donc leurs algorithmes pour prédire les formes de ces protéines, qui sont ensuite comparées à leurs « vraies » structures afin de calculer un score de performance.
Cette année, DeepMind a proposé sa deuxième version d’AlphaFold, qui est sensiblement différente de la première itération en termes d’architecture. Cette fois-ci, les chercheurs ont tiré parti des nouveaux mécanismes “d’attention » et de “transformers” en deep learning combinés aux idées réussies de la première version. L’algorithme a été entrainé sur un jeu de données publique, issu la Protein Data Bank, contenant environ 170 000 séquences de protéines et leurs structures correspondantes (c’est-à-dire les étiquettes dans le cadre d’un apprentissage supervisé). L’entrainement a été effectuée sur un matériel informatique comprenant 122 TPUs de 3e génération (puces spéciales conçues sur mesure pour accélérer l’apprentissage des réseaux de neurones), ce qui équivaut à 100-200 GPUs modernes et a duré quelques semaines. L’entreprise a refusé de révéler le coût de l’entrainement, mais à titre de référence, Google facture 32 dollars de l’heure pour un seul TPU de 3e génération, ce qui donne environ 690 000 dollars pour une seule semaine. AlphaFold v2 a participé au CASP14 (le concours de cette année), et a remporté la première place pour la deuxième fois consécutive. La deuxième version du programme d’IA a obtenu un score médian de 92,5/100, 90 étant l’équivalent des méthodes expérimentales traditionnelles. En conséquence, les organisateurs du CASP et les chercheurs de DeepMind ont déclaré que le problème du repliement des protéines a été résolu.
Implications
La capacité de comprendre la forme des protéines rapidement et avec précision pourrait révolutionner les sciences de la vie. Comme la structure 3D d’une protéine détermine son rôle et ses fonctions, le fait de prédire le résultat du processus de repliement à l’aide d’un
ordinateur permettrait aux chercheurs de découvrir ce que fait chaque protéine. De ce fait, les chercheurs peuvent par exemple répondre à des questions sur la manière dont les anticorps combattent les virus ou dont l’insuline régule le taux de sucre.
Les structures des protéines jouent également un rôle essentiel dans la conception et la découverte de médicaments et dans la compréhension de la santé et des mécanismes qui sont à l’origine de certaines maladies, comme le cancer. Il peut falloir des années de recherche et des milliards de dollars d’investissements afin de créer un nouveau médicament, ce qui freine inévitablement les efforts de recherche. Prédire la structure des protéines liées à une maladie peut accélérer la recherche et réduire considérablement les
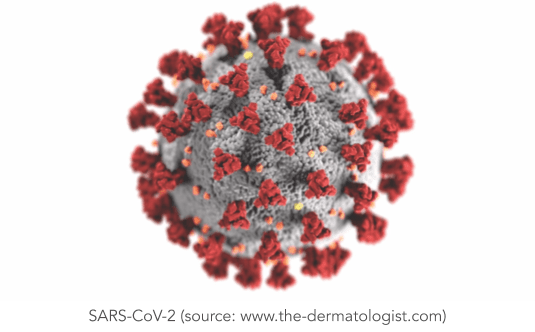
coûts associés. Lorsque le Covid-19 a vu la lumière du jour, nous en savions très peu sur son sujet. Le SRAS-CoV-2, le virus responsable du Covid-19, est composé de 30 types de protéines différentes, dont un tiers est mal connu. Dans ce cadre, les chercheurs se sont intéressés particulièrement à l’étude de l’interaction entre la protéine pointue localisée sur la surface du virus du SRAS-CoV-2 et les récepteurs dans les cellules humaines. Pour faire progresser notre compréhension du virus, l’équipe de recherche derrière AlphaFold a utilisé son programme d’IA pour prédire les structures de quelques protéines sous étudiées associées au virus Corona.
En outre, la connaissance limitée des structures des protéines et le manque de financement ont considérablement entravé les progrès dans la compréhension de certaines maladies tropicales qui ont un impact sur la vie de millions de personnes et entraînent de nombreux décès chaque année. DeepMind a déclaré avoir commencé à travailler avec quelques groupes de recherche afin de concentrer leurs efforts sur certaines de ces maladies.
La résolution du problème du repliement des protéines devrait également ouvrir la voie à de nouvelles opportunités telles que la découverte ”d’enzymes vertes » qui décomposent les déchets plastiques et réduisent ainsi la pollution, le développement de produits agricoles plus nutritifs pour améliorer la santé humaine ou la capture et stockage efficaces du carbone de l’atmosphère.
La prochaine étape ?
AlphaFold v2 est certainement une nouvelle prometteuse pour la communauté scientifique. Toutefois, comme pour tout autre effort de recherche, des améliorations peuvent être envisagées. L’équipe de recherche à l’origine de l’algorithme pense qu’il peut être amélioré davantage pour être encore plus précis. Par ailleurs, le seul point de référence de performance dont nous disposons jusqu’à présent est le score obtenu sur l’échantillon de test fourni par la communauté du CASP. Malgré la résonance de cette réussite, d’autres tests restent nécessaires pour mieux comprendre les limites de ce modèle prédictif, et évaluer avec précision ses capacités de généralisation à des protéines différentes.
Maintenant que le problème est plus ou moins résolu pour les protéines simples, la prochaine étape logique consiste à développer de nouvelles techniques pour prédire la forme des complexes protéiques, qui sont des structures composées de deux protéines ou plus et constituent le fondement de nombreux processus biologiques. Une autre lacune dans la littérature consiste à comprendre comment les protéines interagissent avec d’autres macromolécules telles que les lipides, les glucides ou l’ADN.


